

Building Robust IVDs – Fundamentals of regulated product development (Part 2)
- Mar 1
- 3 min read
Updated: May 11
This is part 2 of a 2-part series.
For part 1, see:

Risk management: a regulatory cornerstone
Risk management is not optional—it is a core requirement of medical device and IVD regulations worldwide. It is defined as the systematic process of identifying, analysing, evaluating, controlling, and monitoring risks associated with a device throughout its lifecycle.

Effective risk management protects:
Patients and users
Healthcare systems
Manufacturers themselves
Why risk management is essential
Robust risk management is driven by:
Ethical responsibility to protect patients
Legal and regulatory obligations
Compliance with international standards
The need to understand real-world use and misuse
Anticipating hazards early allows teams to:
Design safer and more usable products
Prevent failures, recalls, and field actions
Reduce liability and reputational risk
A cross-functional activity
Risk management cannot sit solely within R&D. A comprehensive approach requires input from:
Clinicians and end users
Quality and regulatory teams
Manufacturing and supply chain
Marketing and commercial functions
Each stakeholder brings a unique perspective that strengthens hazard identification and risk control.
Risk Management File
A Risk Management File should be established for each medical device. This file will be subject to review during audits and regulatory submissions and should include the following:
Risk Management Plan
Risk Analysis (hazard identification)
Risk Evaluation (severity, probability, RPN)
Risk Controls (documented evidence)
Determination of Risk Acceptability (signed statement)
Risk Management Reviews
Feedback on Production and Post-Production Risks
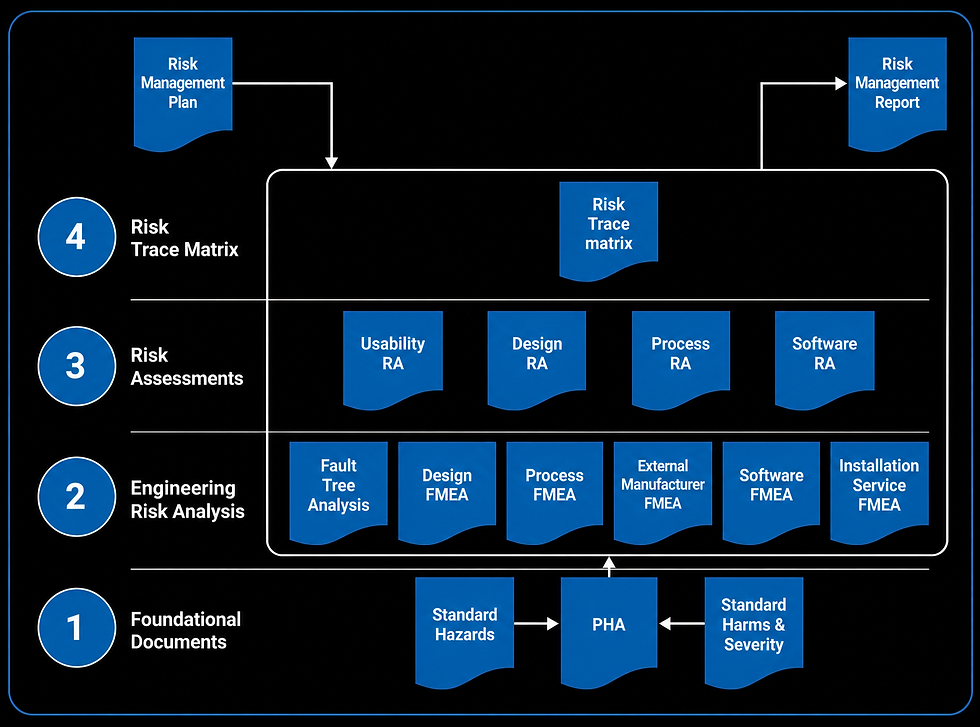
There are many Risk Analysis techniques and guidelines suggest the use of more than one technique to ensure a broad coverage of risks in both normal and failure modes. Common techniques include:
• Preliminary Hazard Analysis (PHA)
• Failure Mode and Effects Analysis (FMEA)
• Fault Tree Analysis (FTA)
• Event Tree Analysis (ETA)
• Hazard and Operability Analysis (HAZOP)
• Hazard Analysis and Critical Control Point (HACCP)
At Device Scope, we help our clients embed risk management into the heart of the development process, ensuring it actively informs design decisions rather than becoming a retrospective documentation exercise.
Standards and guidance
Risk management for IVDs is primarily structured around ISO 14971, supported by additional technical reports and regulatory guidance. Together, these frameworks help manufacturers:
Apply consistent risk concepts
Address cybersecurity and usability
Maintain a complete and traceable risk management file
Relevant references
EN ISO 14971:2019 – Application of Risk Management to Medical Devices
ISO 14971:2019 has been updated to include the new annex A11 2021 – is not considered to be a harmonized standard for the EU In Vitro Diagnostics Regulation (IVDR)
ISO TR 24971:2020 Medical Devices Guidance on the Application of ISO 14971
2020 revision has several updates from 2013 version, including the addition of several helpful new annexes:
Annex D – Risk concepts
Annex F – Risk management for cybersecurity
Annex G – Risk management file
Annex H - IVDs
“Implementation of risk management principles and activities within a Quality Management System”. GHTF/SG3/N15R8:2005.
“Factors to Consider When Making Benefit-Risk Determinations in Medical Device Premarket Approval and De Novo Classifications”, Guidance for Industry and Food and Drug Administration Staff, FDA, 30-Aug-2019
Regulation (EU) MSR 2017/745 and IVDR 2017/746 – Annex I – GSPRs (General Safety and Performance Requirements)
“Applying Human Factors and Usability Engineering to Medical Device", Guidance for Industry and Food and Drug Administration Staff, FDA, 03-Feb-2016
“Recommendations for Clinical Laboratory Improvement Amendments of 1988 (CLIA) Waiver Applications for Manufacturers of In Vitro Diagnostic Devices”, Guidance for Industry and Food and Drug Administration Staff, FDA, 26-Feb-2020
FDA Diagnostic templates for EUA (Emergency Use Authorization) submissions
IVD Product Development – From Concept to Market with Confidence

Design controls in IVD development
Successful IVD development relies on operating under robust design controls, as defined within ISO 13485. These controls structure development activities and ensure that products consistently meet user needs, regulatory requirements, and performance claims.
Key design control elements include:
Design planning
Design inputs and outputs
Design reviews
Verification and validation
Design changes
Structured development models
Design controls are often illustrated using a waterfall model, where each development stage builds on the last. For larger or more complex programmes, a phase-gated project management approach is frequently used.
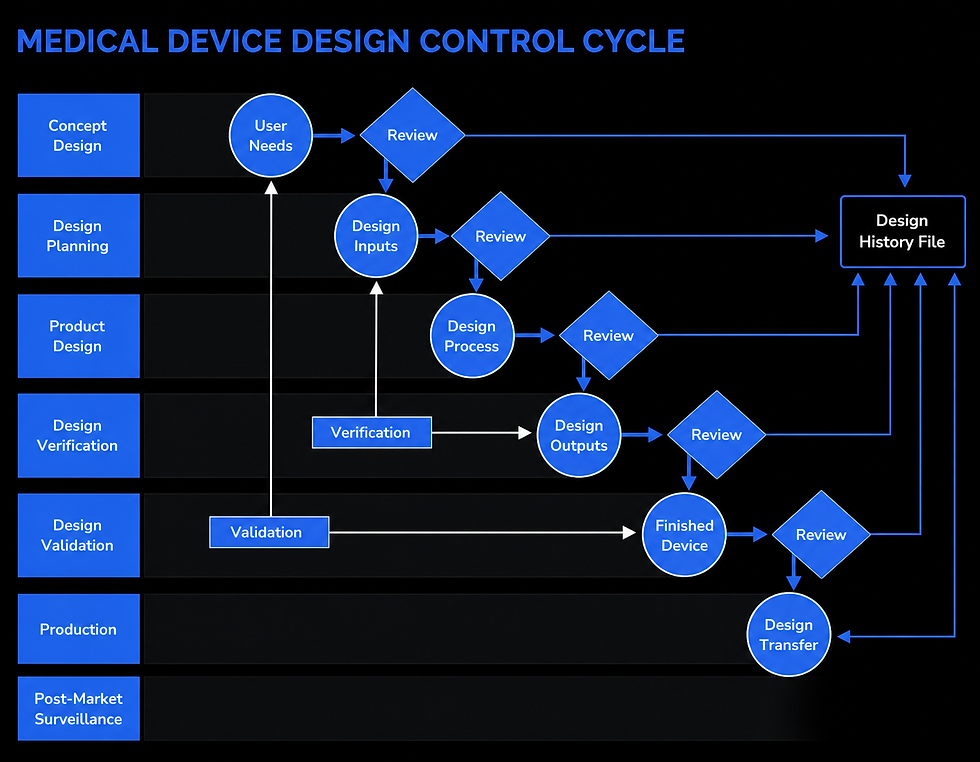
In a phase-gated model:
Development is divided into defined phases
Each phase ends with a formal design review (a “gate”)
Progression is based on meeting predefined criteria

Why phase-gated development works
This approach:
Reduces technical and regulatory risk
Improves cross-team communication
Ensures efficient use of resources
Increases the likelihood of first-time-right development
Device Scope: more than technical delivery
At Device Scope, we support clients not only with cartridge and instrument development work packages, but also with programme leadership—helping teams manage complexity, align stakeholders, and move confidently toward market launch.


Comments